
 |
 |

 |
 |
 |
 |
 |
|
|
 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
The Virtual Ribosome
The role of the ribosome in the protein folding process is a subject of controversial discussions. On the one hand denatured proteins can regain their native conformation in vitro, on the other hand this ability decreases with growing molecular weight. In addition, the folding time of a protein is in the same order of magnitude as the synthesis time at the ribosome, which makes the start of folding during chain elongation a reasonable assumption. Intuitively, one would expect that co-translational folding becomes increasingly important with growing chain length, and indeed it has been experimentally proven several times for multi-domain proteins, that simply do not fold properly without a ribosome [1].
YASARA can be used to simulate the folding of a protein at the ribosome (fig. 1). However, the aim is not a realistic reproduction with explicit water molecules, but the efficient minimization of a complex energy function in vacuo, taking into account the ribosomal asymmetry. In addition to the YASARA NOVA Force Field, the energy function considers predefined atomic distances (e.g. NOESY distance restraints and/or secondary structure predictions), as well as the effects of the solvent in form of hydrophobic pseudo forces [2].
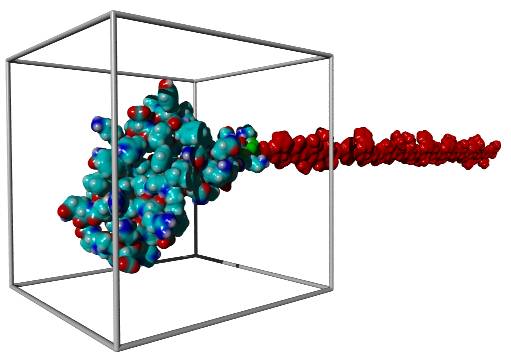
Fig. 1: A virtual ribosome. The polypeptide chain is moved step by step into the simulation box, where the energy function is minimized using molecular dynamics combined with simulated annealing.
References:
1. Frydman, J. et al. (1999) Nat.Struct.Biol.6(7),697-705
2. Casari, G., Sippl, M.J. (1992) J.Mol.Biol. 224, 725-732