








Protein side-chain modeling in YASARA
YASARA Structure routinely predicts protein side-chain conformations ('rotamers'), ranging from single point mutations to complete homology models:
- Protein side-chain rotamers are strongly influenced by the backbone conformation[1]. YASARA therefore uses the φ and ψ dihedral angles to extract the preferred rotamers and the associated probabilities from a backbone dependent rotamer library[2].
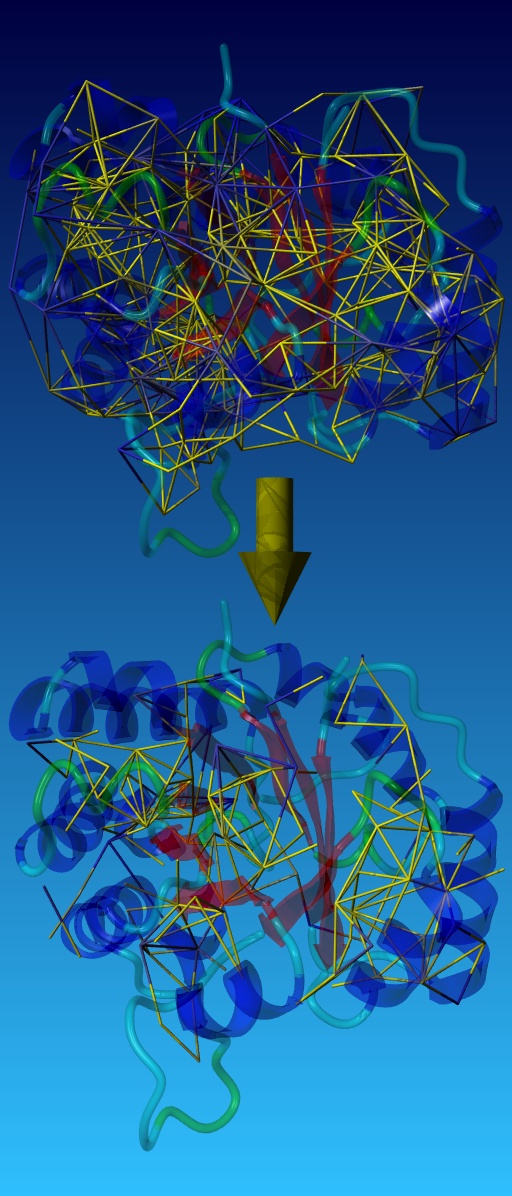
- Based on the available rotamers, YASARA determines which side-chains can potentially influence each other and builds a graph of interacting residues (top half of the figure on right).
- Using a fast repulsive energy function and dead-end elimination, the complexity of the graph is reduced significantly (bottom half of the figure on the right). Finally, the remaining graph portions are further broken down to sets of biconnected components, which are small enough to be solved by exhaustive enumeration[3].
- The repulsive energy function used in the previous step works very well for those side-chains that interact mainly via repulsive Van der Waals forces, i.e. the hydrophobic residues in the core. To capture the complex electrostatic interactions on the surface, YASARA performs another optimization round that explicitly considers electrostatic and solvation effects, as well as subtle packing preferences (via multi-dimensional knowledge based potentials) and deviations from the idealized rotamer geometry.
- pH dependent side-chain interactions with ligands are fully accounted for, as are unusual amino acids.
- Backbone dependent side-chain conformations can alternatively be extracted directly from a non-redundant subset of the PDB.
R E F E R E N C E S
[1] The use of position-specific rotamers in model building by homology
Chinea G, Padron G, Hooft RW, Sander C, Vriend G (1995) Proteins 23, 415-421
[2] Bayesian statistical analysis of protein side-chain rotamer preferences
Dunbrack RL Jr., Cohen FE (1997) Protein Sci. 6,1661-1681
[3] A graph-theory algorithm for rapid protein side-chain prediction
Canutescu AA, Shelenkov AA and Dunbrack RL Jr. (2003), Protein Sci. 12,2001-2014.