








The YASARA PDB_REDO interface
PDB_REDO is a databank of updated and optimized X-ray structure models developed by Dr. Robbie Joosten[1]. PDB_REDO re-refines the X-ray structures in the PDB using the original raw data (reflection files) with state-of-the-art technology to arrive at more accurate structure models (which typically have a lower R-factor and better structure quality indicators).
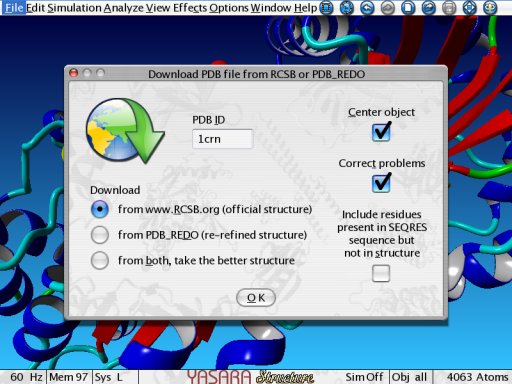
PDB_REDO is tightly integrated in YASARA. When you select to download a PDB file from the Internet (File > Load > PDB file from Internet), you can choose between the original PDB file from the RCSB, the re-refined file from PDB_REDO, or the better of the two. In the latter case, YASARA downloads both files, and picks the one with the better WHAT IF structure validation results[2]. You can also download PDB_REDO files from your Yanaconda macros or Python scripts using the LoadPDB command.
R E F E R E N C E S
[1] PDB_REDO: automated re-refinement of X-ray structure models in the PDB
Joosten RP, Salzemann J, Bloch V, Stockinger H, Berglund A-C, Blanchet C, Bongcam-Rudloff E, Combet C, Da Costa AL, Deleage G, Diarena M, Fabbretti R, Fettahi G, Flegel V, Gisel A, Kasam V, Kervinen T, Korpelainen E, Mattila K, Pagni M, Reichstadt M, Breton V, Tickle IJ, Vriend G (2009) J. Appl. Cryst. 42:376-384
[2] Errors in protein structures
Hooft RWW, Vriend G, Sander C, Abola EE (1996) Nature 381, 272