
 |
 |

 |
 |
 |
 |
 |
|
|
 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
Searching for the DNA binding site of proteins
Control of gene regulation represents a major cornerstone in the experimental repertoire of modern molecular biology. The ability to express genes only when desired is for example used in "Runaway - Plasmids", where a temperature-sensitive Lambda- repressor (CI 857) blocks the transcription of the plasmid's replication genes. If one increases the temperature to 42°C, CI 857 is deactivated, expression of replication genes starts and the plasmid copy number is increased. Controlling gene regulation also offers a broad range of therapeutic applications. Since large proteins cannot penetrate the cell membrane, one designs usually short peptides or other small organic molecules, that bind in the minor groove of the DNA [1]. In order to bind DNA specifically, a protein must detect its target sequence accurately (otherwise e.g. the expression of the endonuclease EcoRI would have fatal consequences for E.coli).
|
Essentially two mechanisms are available: 1) recognition of the sequence- dependent helix structure: the stacking of the base pairs usually does not stick to the ideal B- DNA form. Rotations and shifts (slide, twist and roll), as well as a certain influence of the sequence on the backbone conformation are known to occur [2]. In many cases the DNA must even deform strongly to match the entire binding-site of the protein. Whether such serious structural changes are energetically possible, depends again on the base- sequence. In general, this recognition method is however too inaccurate to account for highly specific interactions. Therefore a second, more differentiated method was established during evolution: 2) direct recognition of the DNA-sequence by hydrogen bond formation and hydrophobic contacts between bases and amino acids. If one looks at the crystal structures of protein / DNA-complexes in the PDB, then this direct read-out mechanism is involved in practically all the cases of specific binding.
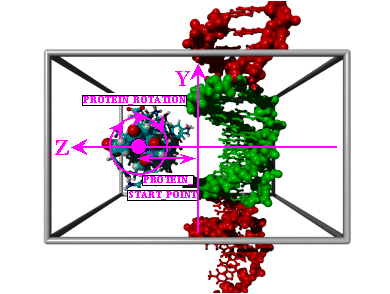
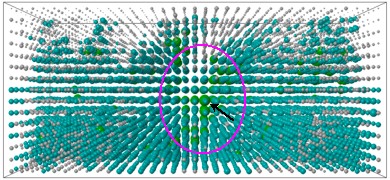
One possible application of the docking module YASARA DOCK is the prediction of these specific protein/ DNA-interactions. If the structure of the regulatory protein and the sequence of the operator region where it binds the DNA are known, YASARA can systematically scan the conformational space by running all-atom molecular dynamics simulations of the docking event in thousands of different orientations. This becomes especially feasible in the case of alpha helices binding in the major DNA groove, as just three degrees of freedom need to be searched: 1) The position along the DNA main axis (fig.1), 2) the shift of the alpha- helix (fig.1) and 3) the rotation of the alpha- helix (fig.2). As a positive control, YASARA tried to determine the binding site of the a1-alpha2 - protein complex from Saccharomyces cerevisiae. In diploid yeast cells with active MATa and MATalpha loci, the two proteins a1 and alpha2 form a heterodimer with various regulation functions. Among other things they repress the transcription of the genes HO (change of mating type) and RME1 (repressor of meiosis). The DNA-binding motif belongs to the well known "helix-turn-helix" class, and one of the two binding helices involved (fig.3) was chosen for the experiment. The alpha- helix was docked to the DNA in 5776 different orientations along a 20bp DNA-fragment, that contained the same sequence as the DNA in the experimentally determined crystal structure. The result is shown in fig.4. For each of the 5776 docked and energy- minimized complexes, the specific binding energy is indicated as a ball. The larger and greener the ball, the higher the binding energy. As three degrees of freedom were used, the resulting diagram is also three dimensional, with every axis corresponding to one degree of freedom. Balls close in space thus belong to similar relative orientations of protein and DNA. It is immediately apparent that good solutions (large green balls) form clusters in conformational space, and the conformation with the true, native binding site and also the correct hydrogen bonding pattern (marked with an arrow) even corresponds to the largest ball in the largest cluster. If clustering is not taken into account, and energies alone are evaluated, the native-like conformation is ranked third of 5776. There are thus two false positives that score better than the native-like conformation. When the method has been improved to reliably sort out these false positives, YASARA DOCK will become a publicly available module of YASARA Structure. References: 1. Kielkopf, C.L., White, S., Szewczyk, J.W., Turner, J.M., Baird, E.E., Dervan, P.B., Rees, D.C (1998) Science 282, 111-115 |
 |
|
| Fig.1: Docking of an alpha helix in the major DNA groove, part 1. Two of the three degrees of freedom are shown: The position along the DNA main axis (DNA_AXIS_SHIFT) and the shift of the alpha helix within the major groove (HORIZONTAL_SHIFT) . | ||
 |
||
| Fig.2: Docking of an alpha helix in the major DNA groove, part 2. The third degree of freedom, the rotation of the alpha helix about its main axis (PROTEIN_ROTATION), is shown. | ||
 |
||
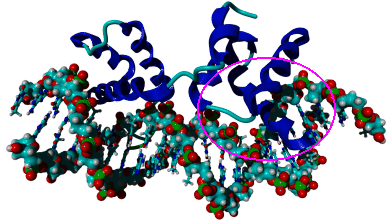
| Fig.3: Crystal structure of the a1- alpha2 - heterodimer from Saccharomyces cerevisiae , complexed with synthetic DNA (PDB entry 1AKH). The 26 residue long DNA binding alpha helix of the a1 protein that was used in this experiment is marked with a magenta circle. | ||
 |
||
| Fig.4: Three-dimensional representation of a DNA-docking experiment. Each of the 5776 tested conformations is represented by a ball, the larger and greener a ball, the higher the specific binding energy of that conformation. The largest green ball (arrow) in the largest green "hot spot" (magenta circle) is the native-like complex (same recognition site, same hydrogen bonding pattern). |