








YASARA CASP8 Results
|
CASP ('Critical Assessment of Structure Prediction') is a biennial evaluation of today's many approaches to protein structure prediction, organized by the Prediction Center since 1994. During each CASP season (lasting ~4 months), about 200 research groups try to predict the structures of ~100 proteins (the CASP targets), applying all combinations of prediction methods, ranging from experimental homegrown ones to well established molecular modeling packages. The target sequences are provided to CASP by structural biology labs (mostly structural genomics projects) just before the corresponding structures are solved. The predictions are thus real 'blind predictions', which makes CASP a unique opportunity to judge if a certain method really works, or is mainly based on wishful thinking and a lot of advertisement. Interestingly, the most expensive solutions hardly ever perform well at CASP, showing that users of molecular modeling software must be careful not to waste their resources. |
CASP8 Refinement Section - The last mile of the protein folding problem
One of YASARA's main functions has always been the atomistic simulation of proteins. Unfortunately today's computers are much too slow to run a full molecular dynamics simulation of a folding protein for all but the simplest peptides. CASP came to the rescue by introducing the refinement section: predictors are provided with the best model submitted for a certain target (created with any other method), and can then use their high-resolution refinement simulations to improve the model and move it closer to the target (the 'last mile of the protein folding problem'). As it turns out, this problem is already hard enough: while it is trivial to shake a protein around by molecular dynamics simulation, most of the shaking goes in the wrong direction, usually due to the limited accuracy of today's empirical force fields. Given the difficulty of the problem, we are happy to report that YASARA's molecular dynamics simulations won 3 of the 12 refinement targets, and nobody else won more targets than YASARA (using the CASP Model_1-only ranking for targets TR429, TR454 and TR469, see table 1 below). We congratulate the Baker Lab for also having won three targets (others won at most one target). Since their Monte-Carlo based Rosetta program has regularly disqualified molecular dynamics based methods during the previous CASPs, it is great news to see that eight years of research on increasing the accuracy of YASARA's force fields[1,2] has made molecular dynamics competitive again, at least in the refinement section. Predictions were made fully automatically without human intervention. Additional help came from WHAT IF in the Twinset to provide a second opinion on model quality, and from CONCOORD to speed up sampling under certain conditions. It should be noted that despite these successes, there still exists no method today that can consistently improve every single model, some still go into the wrong direction. Work to open this bottleneck is in progress. |

|
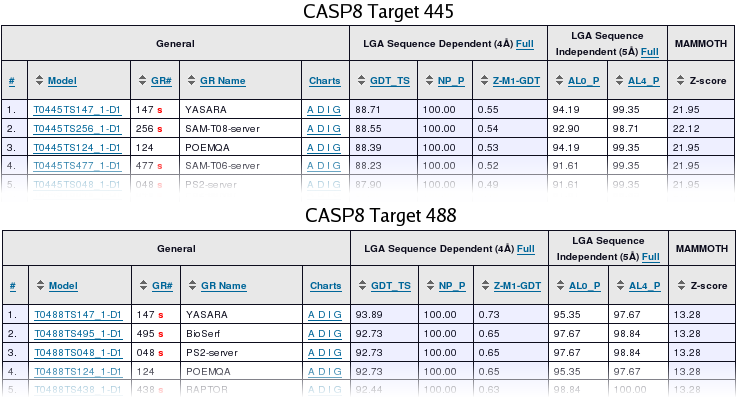
CASP8 Homology Modeling Section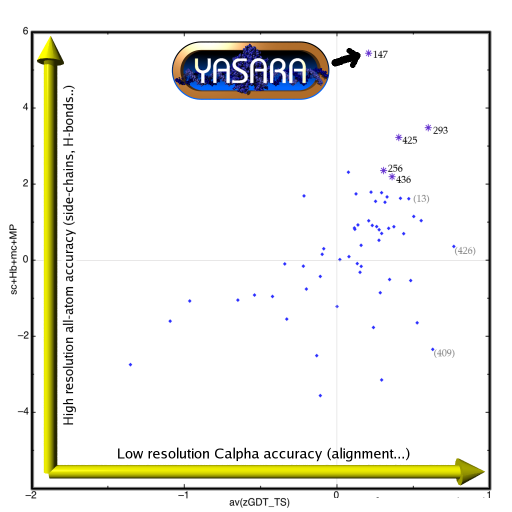 YASARA's homology modeling module was registered as a CASP server and thus had to provide predictions within three days after target release, fully automatically and without human intervention. Models were submitted for 61 targets, whenever PSI-BLAST found a useful homology modeling template and the resulting model was of acceptable quality. YASARA won two homology modeling targets (T445 and T488, see table 2 below), and more importantly, was overall ranked first in the category 'high-resolution server accuracy' by the assessors. This is shown along the vertical axis in Figure 3 on the right. Since it involves especially side-chains and hydrogen bonds, YASARA obviously benefited from its side-chain modeling and high-resolution refinement algorithms. There is still room for improvement along the horizontal low-resolution C-alpha accuracy axis, requiring tuned alignments and improved fusion of multiple templates. Again, this topic is currently being worked on. Additional help came from the PDB-Redo database, which supplied re-refined templates. |
 |
|
The YASARA results have been published in the CASP8 special issue of the journal Proteins: Structure, Function and Bioinformatics, including the YASARA force field with knowledge-based dihedral potentials, that has been optimized to yield stable energy minima close to native X-ray structures. YASARA's homology modeling module is also outlined briefly. The journal cover shows how the various dihedrals in an arginine residue are combined by YASARA's multi-dimensional potentials. 
Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8 Krieger E, Joo K, Lee J, Lee J, Raman S, Thompson J, Tyka M, Baker D, Karplus K Proteins. 2009;77 Suppl 9:114-22 R E F E R E N C E S |